Why do MDS clusters when we already did PCA-based clustering for this data?
You guys probably know about Dienekes' Clusters Galore approach. The way it works is that varying the number of MDS dimensions used you compute the number of clusters inferred (done using Mclust) and use the number of MDS dimensions which give you the maximum number of clusters.
This sounded a little unsatisfactory for me. So I ran an experiment. I computed 100 MDS dimensions for the samples in this dataset which includes South Asians from Reference II as well as 38 Harappa participants. Then I kept 2,3,4,...,100 dimensions and ran NNClean (to get initial noise/outlier estimate) and Mclust on them.
This first graph shows the number of outliers NNclean computed from 586 samples.

Things go crazy with NNclean when 64 or more MDS dimensions are retained since it considers most of the samples to be noise then.
Now let's look at the number of outliers identified after Mclust's clustering procedure.

This shows us that probably somewhere between 8 and 65 MDS dimensions might be useful to keep.
Finally, a plot of the number of clusters inferred by Mclust versus the number of MDS dimensions used.

There are two big jumps here to consider. One is around 12 MDS dimensions and the other after 52. So we are looking at an optimum number of MDS dimensions between 12 and 52. However, in that range, the number of clusters computed is fairly noisy between 18 and 26. The only pattern I can discern with some smoothed fitting is that we should likely be looking at somewhere between 20 and 30 MDS dimensions.

But why choose the maximum number of clusters (26 clusters when 24 MDS dimensions are kept)? That could be the result of noise too.
Is there some other way to figure out what are the significant number of MDS dimensions to keep for population structure? It turns out there is. Patterson, Price and Reich proposed Tracy-Widom statistics for Principal Component Analysis in their paper "Population Structure and Eigenanalysis". We also know that the MDS analysis we are performing is the classical metric MDS which is in some ways equivalent to a PCA. Looking at the Tracy Widom stats then, we see that about 25 eigenvalues are significant. Thus, keeping 24 MDS dimensions to maximum the number of clusters seems defensible.
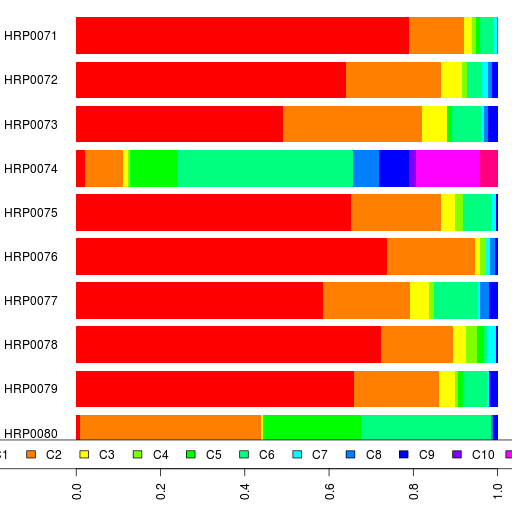
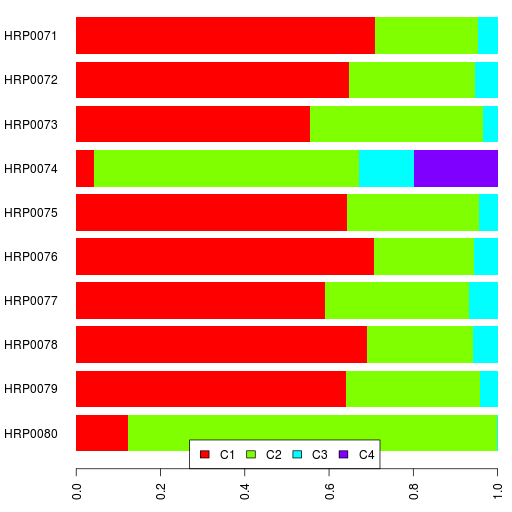
Finally, here are the clustering results.










{kind=link}
{kind=link}
Recent Comments